The autonomic nervous system confuses me
The difference between anti-muscarinic and anti-cholinergic toxicity, organophosphate poisoning, whatever the hell atropine is doing, the concept of a “nicotinic AchR” — none of these made any sense to my poor brain as a medical student. On an anesthesiology rotation I sat down with one of my attendings at lunch and asked him to explain these things to me, not so I could write a disastrous blog post years later, but so that I could understand what was going on with the neo/glyco reversal combination. He spent 30 minutes talking to me. He drew multiple diagrams. I had no idea what was going on at the end of that lunch.
So, here goes nothing: the autonomic nervous system, and its receptors, for those of us desperately holding onto our last remaining synapse. Or neuron.
Some general anatomical tomfoolery
The autonomic nervous system is broadly described as having three parts: the sympathetic nervous system, the parasympathetic nervous system, and the enteric nervous system. The sympathetic nervous system corresponding roughly to a state of physiologic acceleration, and the parasympathetic to a state of physiologic deceleration. The enteric nervous system is its own strange beast, being arguably the oldest evolutionarily of all of our nervous systems, and scares me enough that I don’t particularly want to touch it today. My apologies to Julia Kaltschmidt, who certainly has no idea who I am. Thank the lord.
Ganglia shmanglia
The sympathetic and parasympathetic nervous systems consist of a set of pre-ganglionic neurons that run from the brain to a set of ganglia, and then synapse upon a set of post-ganglionic neurons that are made to do the dirty work of actually telling organs what to do. In this sense the ganglia can be thought of as signal integration centers, that parse “higher-order” commands from the brain into more concrete directions for particular organs. Somewhere out there, a neuroscientist has just collapsed from an aneurysm after the completion of that sentence.
Parasympathetic ganglia tend to lie close to their target organs; for example, there are named parasympathetic ganglia for several organs (ciliary ganglion for the eye, pterygopalatine ganglion for the lacrimal gland) and ganglia near their target organs that can be identified downstream of various plexuses. In contrast, sympathetic ganglia tend to either be within the spinal cord, with post-ganglionic fibers innervating multiple target organs, although there are exceptions to this rule. A quick look comparing the Wikipedia figures for the sympathetic and parasympathetic nervous systems may prove useful – preganglionic fibers are in red, while postganglionic fibers are in black. The sympathetic nervous system has more black stuff near target organs; the parasympathetic nervous system, more red stuff.
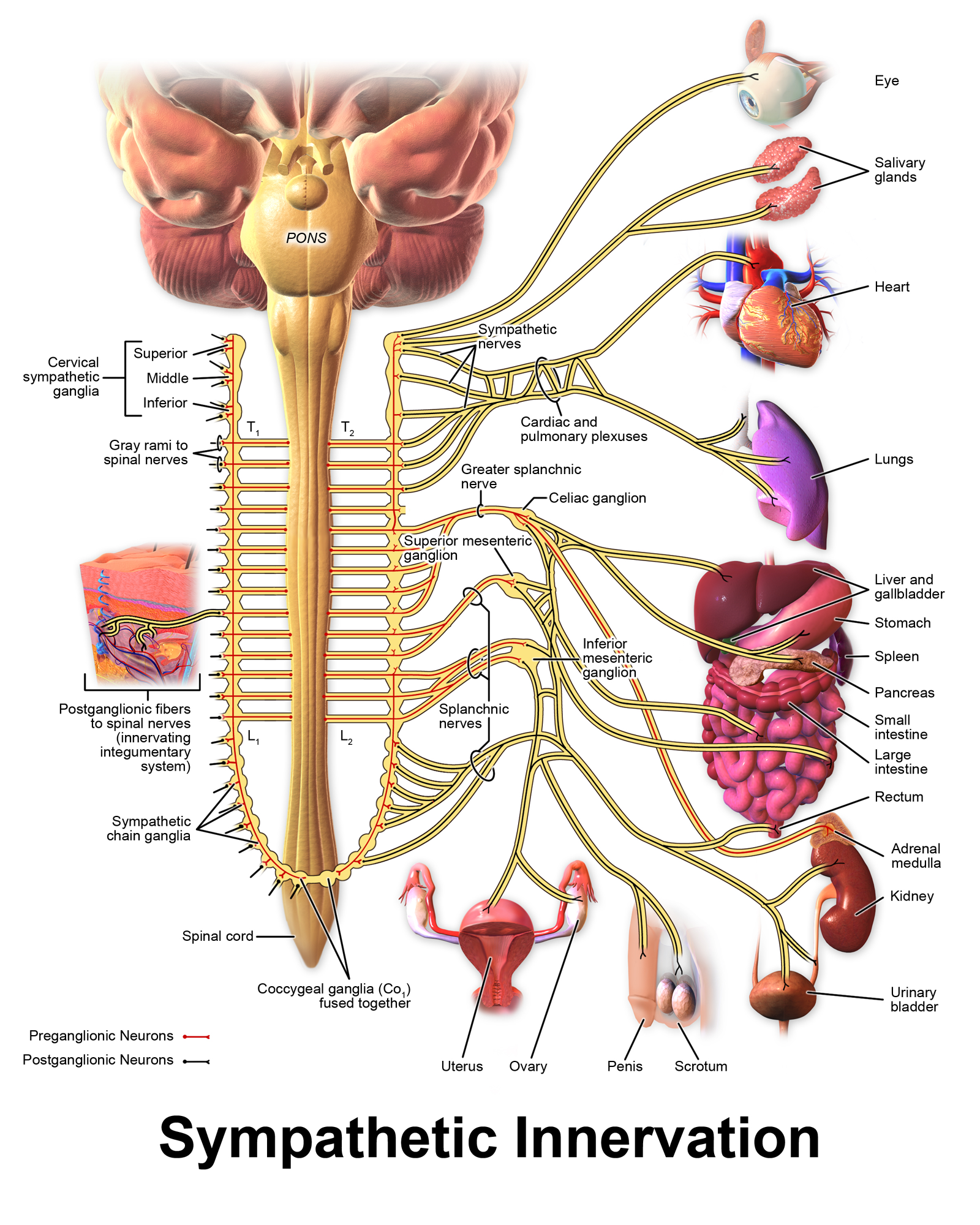
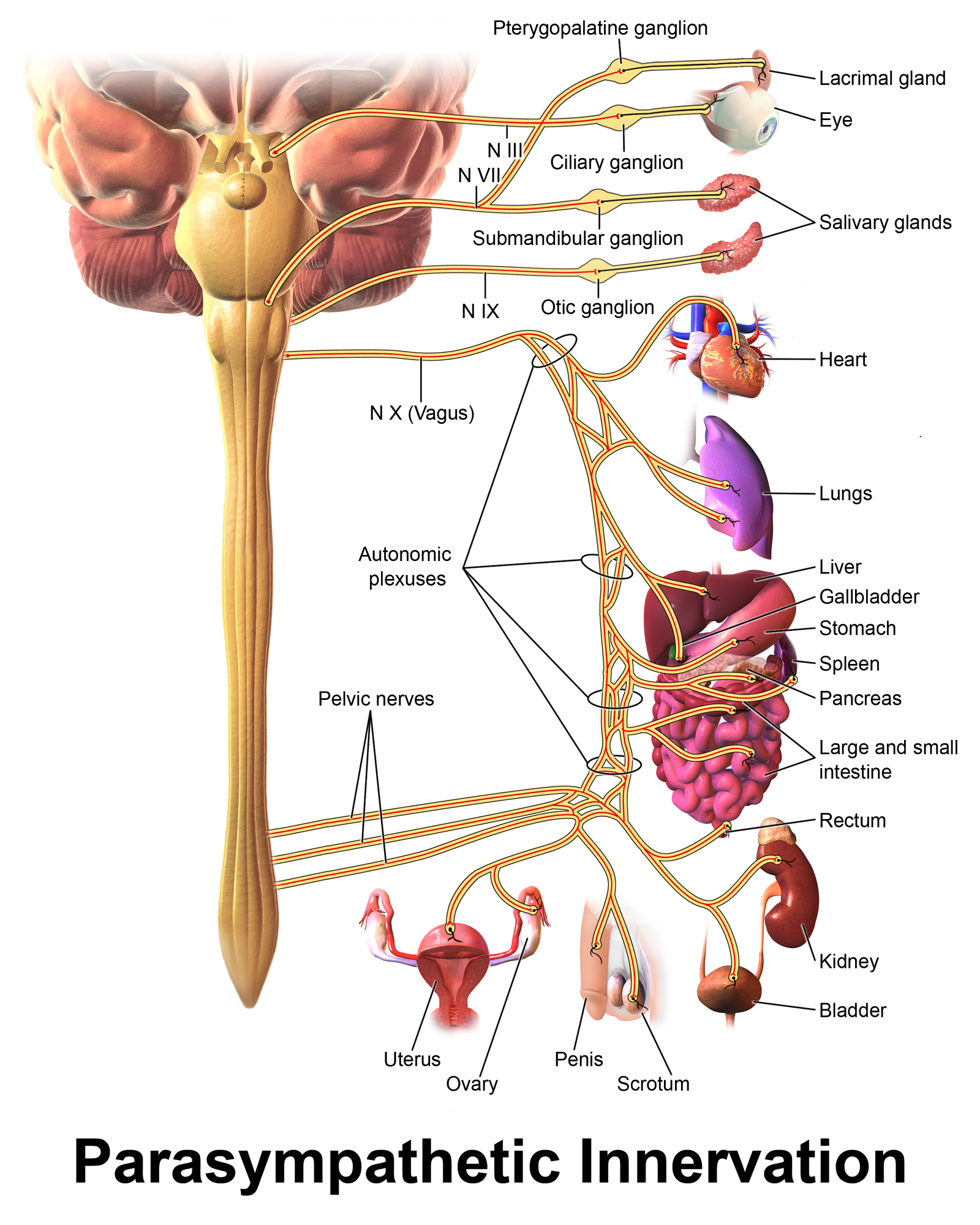
Neurotransmitters aplenty
This gives us 2 sets of synapses for the sympathetic and parasympathetic nervous systems: pre-ganglionic and post-ganglionic. Understanding these synapses clears up a lot of the anti-muscarinic anti-cholinergic mumbo jumbo that confused me throughout the litany of DUMBELS/SLUDGE mnemonics I endured in medical school.
Everything preganglionic uses acetylcholine (which was used for cell-cell communication since before the development of neurons themselves) – and, specifically, nicotinic acetylcholine receptors. They’re called nicotinic because (shocker) they bind nicotine, and they act as ligand-gated ion channels. Postganglionic parasympathetic synapses use muscarinic acetylcholine receptors (named because they bind muscarine more than nicontine), which act as GPCRs (more on that in a moment). Postganglionic sympathetic synapses mostly use norepinephrine as their signaling molecule, which binds
These muscarinic receptors, in turn, have subtypes – 5 of them, to be precise. Subtypes M1, M3, and M5 use G
On the sympathetic side, your sympathetic receptors are organized as follows:
Here’s a little ol’ table with all this from here11. Of course, in full disclosure: I hate tables, can never memorize them, but I suppose it’s useful as a reference.:
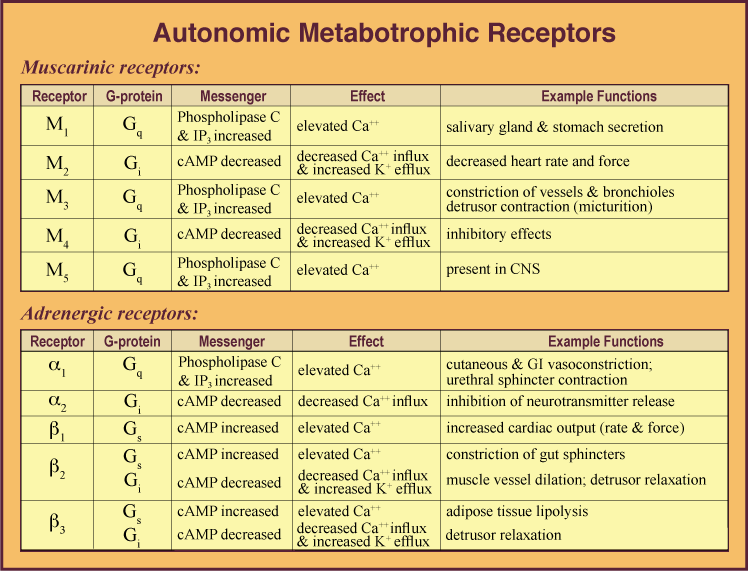
So, post-ganglionically-speaking, we have a bunch of scattered GPCRs triggered by Ach or norepinephrine. Why do we care?
GPCRs
It may be useful at this time to remind yourself (read: myself) how GPCRs work. GPCRs are receptors comprised of 7
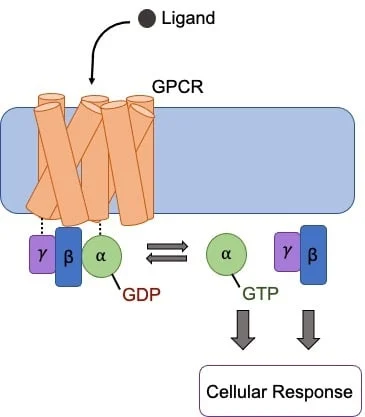
The downstream effect of G
Why do we care about this in the context of sympathetic/parasympathetic stimulation? Well, it helps explain why things happen. For example:
Toxicity
So, how does this help us (read: me) understand muscarinic vs cholinergic toxicity? Well, muscarinic toxicity can be thought of as overactivation of your M1-5 receptors: you get bradycardia, bronchoconstriction, and lots o’ secretions. Then, cholinergic toxicity is this plus the effects of the nicotinic receptors – neuromuscular junction shenanigans, mixed sympathetic/parasympathetic findings, CNS symptoms. Organophosphate poisoning is cholinergic because of inhibition of acetylcholinesterase; muscarinic toxicity is more often seen in muscarine overdose (lol), and pilocarpine/bethanechol (M3 agonists) toxicity.
From a treatment perspective, atropine is a competitive and reversible muscarinic antagonist – which means, on its own, it will not treat the full range of symptoms from cholinergic toxicity. In contrast, neostigmine is a acetylcholinesterase inhibitor that does NOT cross the blood-brain barrier (in contrast, physostigmine does enter the CNS; it lacks neo’s quaternary nitrogen atom). We then combine that with glycopyrrolate, a competitive muscarinic antagonist (like atropine!) to suppress the muscarinic toxicity while letting the neo help rebuild acetylcholine at the neuromuscular junction. We use glyco rather than atropine because a) atropine enters the CNS, which we’d like to avoid), and b) it lasts longer, so its kinetics are better matched to neostigmine. In contrast, atropine is a reasonable match for the alternative acetylcholinesterase inhibitor edrophonium.